|
XI. MALADIES DU MUSCLE ET DE LA SYNAPSE NEURO-MUSCULAIRE
A. GENERALITES
Les muscles striés sont commandés par les neurones moteurs au niveau de la plaque motrice : cette dernière est la synapse neuro-musculaire (neuromédiateur = acétylcholine).
Les maladies autonomes sans caractère inflammatoire du muscle sont appelées myopathies : il existe des myopathies évolutives, maladies souvent graves. D'autres myopathies sont acquises dès la naissance et n'évoluent
pas (myopathies congénitales).
La connaissance des facteurs génétiques de ces maladies a grandement bénéficié des progrès de la génétique. De très nombreux sites de mutation chromosomique ont été mis en évidence, malheureusement pour le moment
cela n’apporte rien aux possibilités thérapeutiques.
B. LA MYASTHENIE
Maladie auto-immune de la synapse neuro-musculaire, décrite dès 1672 par Thomas Willis. Erb (1879), Goldflam (1891).
Pic femmes avant 20 ans, Hommes après 60 ans. Fréquence environ 1/40.000 habitants ?
Clinique : le phénomène myasthénique, déclenché par l'effort musculaire. Amélioré par la prostigmine. Aggravé par le curare et de nombreux médicaments (crises respiratoires).
L’examen neurologique est normal.
Signes oculaires fréquents (> 90%). troubles de la phonation (81%), mastication, déglutition, face. Tous ces signes ont pour caractéristique leur AGGRAVATION en cours de journée ou bien par l’effort musculaire.
Evolution chronique , capricieuse, prolongée. Poussées sans cause apparente. Rémissions. Gravité des poussées respiratoires.
Il existe une classification selon la gravité de la myasthénie (classification d’Osserman).
Les “crises cholinergiques” sont liées à un excès de traitement (diarrhée, pâleur, sueurs, hypersalivation,...)
Examens complémentaires :
Les bilans biologiques de routine sont normaux.
L’électromyogramme, est le seul examen paraclinique d’orientation permettant une étude de la jonction neuro-musculaire. Chez les sujets myasthéniques, il existe en stimulation répétitive à basse fréquence (3 par
seconde) une diminution de l’amplitude de la réponse musculaire. C’est la “cupule myasthénique”. Ces anomalies se corrigent après injection intraveineuse de prostigmine.
L’exploration fonctionnelle respiratoire peut montrer une insuffisance qui elle aussi dépend de la journée.
Biologiquement on trouve des anticorps anti-récepteurs à l’acétylcholine de façon pratiquement constante dans le sang. Il peut aussi exister d’autres anomalies immunitaires (anticorps anti-muscle strié et
anti-thymus).
Radiographies du médiastin. Importance de la recherche de Thymome.
Formes trompeuses : oculaires, distales, amyotrophiques, respiratoires.
Chez le nouveau né il existe une myasthénie CONGENITALE, transmissible. Les mères ne sont pas myasthéniques, il n’y a pas d’anticorps anti-récepteurs à l’acétylcholine dans le sang.
Parfois association avec épilepsie, affections thyroïdiennes, hémopathies (anémie), Lupus, polyarthrite...
Le traitement de la myasthénie utilise les médicaments inhibant la dégradation de l’acétylcholine au niveau de la synapse neuro-musculaire (médicaments anti-cholinestérasiques): PROSTIGMINE, MESTINON, MYTELASE.
L'infirmier doit se méfier des myasthéniques, très sensibles à de nombreuses médications (somnifères, tranquillisants). Dans le doute, en raison de risques respiratoires importants, ne pas donner le traitement sans
confirmation. A l'inverse, un malade aggravé par ce type de médicaments peut être myasthénique. CHEZ LE MYASTHENIQUE UNE ERREUR THERAPEUTIQUE PEUT ETRE FATALE (exemple: prescription inappropriée de somnifères). Les
médicaments formellement contre-indiqués sont nombreux: certains antibiotiques (AMINOSIDES), la QUININE, le DIHYDAN, les bêta-bloquants, ... Tous les neuroleptiques ou tranquillisants sont très dangereux.
C. LE SYNDROME DE LAMBERT-EATON
Décrit en 1956 par Lambert, Eaton et Rooke sur l’étude de 6 cas dont 5 étaient atteints d’affections cancéreuses. Environ 1 cas pour 100 myasthénies.
Syndrome myasthéniforme habituellement paranéoplasique, habituellement en rapport avec les tumeurs pulmonaires.
Cliniquement se traduit par une faiblesse générale. Amélioration paradoxale par l'effort musculaire.
Localisation préférentielle aux racines des membres.
Nécessité pour le diagnostic de l'électromyogramme effectué dans des conditions très particulières (tétanisation). Est souvent à l'origine de la découverte de la tumeur primitive.
D. DYSTROPHIES MUSCULAIRES PROGRESSIVES (MYOPATHIES)
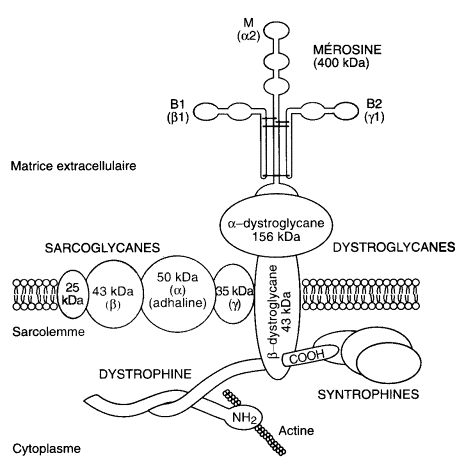
La classification a évolué en raison des connaissances génétiques. D’autre part on connaît mieux la structure du complexe protéique reliant les protéines contractiles intracytoplasmiques liées à l’actine et la
matrice extra-cellulaire de la lame basale qui contient la dystrophine et d’autres protéines de structure.
1. DYSTROPHIES MUSCULAIRES PROGRESSIVES
Importance de l'histoire familiale (génétique).
Aspect EMG particulier.
Enzymes musculaires, biopsie musculaire.
2. DYSTROPHIES LIEES AU GENE DMD
Les recherches ont abouti à la mise en évidence du gêne d’une protéine de structure du muscle, la DYSTROPHINE, sur le bras court du chromosome X.
2.1. La maladie de DUCHENNE DE BOULOGNE.
Maladie de Duchenne (DMD) (1868) : 2,8/100.000. Ne touche que les individus de sexe mâle. Début précoce, évolution progressive. Souvent hypertrophie des mollets. Pronostic défavorable.
La phase préclinique semble se constituer dans la période anté-natale. Les lésions existeraient déjà chez le
foetus. Le début des troubles de la marche se fait vers 3 à 6 ans: chutes, difficultés à monter un escalier. Ensuite se constitue une hypertrophie musculaire vers 5 à 6 ans. Entre 6 et 10 ans les chutes sont
aggravées. Vers 10 ans la marche n’est plus possible. La paralysie est pratiquement totale avant l’âge de 20 ans.
L’atteinte myocardique est constante, souvent précoce. Les problèmes respiratoires sont en général la cause du décès. Un tiers des enfants présente un retard mental.
2.2. La maladie de BECKER.
Formes bénignes : type Becker (début entre 2 et 45 ans, le plus souvent entre 4 et 19 ans). L’évolution est peu invalidante avec une incapacité seulement vers l’âge de 30 ans.
2.3. Examens complémentaires.
L’anomalie majeure est l’élévation des taux d’enzymes musculaires dans le sang (Créatine Phosphokinase CPK). Des auto-anticorps sont souvent présents (anti muscles lisses et striés).
En électromyographie les vitesses de conduction sont normales et les aspects en détection comportent des potentiels d’unités motrices fins et peu amples avec recrutement précoce à l’effort peu intense.
La biopsie musculaire objective l’atrophie des fibres musculaires.
3. LA MALADIE DE STEINERT (Myotonie dystrophique)
1909 (Steinert). C’est la plus fréquente des dystrophies musculaires de l’adulte: 5/100 000. L’hérédité est autosomique dominante avec début de plus en plus précoce de génération en génération.
Sévérité très variable. Evolutivité lente.
Atteinte des muscles de la face, du cou et des segments distaux des membres. ATROPHIE musculaire. Cette atrophie s’accompagne d’une MYOTONIE qui prédomine au niveau des mains. Elle est aggravée par le froid.
Les manifestations cardiaques sont très fréquentes, surtout des troubles de conduction. Il existe un risque de mort subite.
Les troubles respiratoires sont fréquents.
Les manifestations endocriniennes sont constantes: hypogonadisme, stérilité, diminution de la libido, impuissance. Souvent un diabète de type II.
La cataracte est pratiquement constante après 40 ans.
L’hypersomnie est fréquente. La calvitie s’observe chez 80% des hommes.
4. LES MYOTONIES NON DYSTROPHIQUES.
Groupe de maladies héréditaires souvent autosomiques dominantes. Il s’agit d’un dysfonctionnement des canaux ioniques membranaires.
4.1. Canalopathies chlore.
Myotonie congénitale de transmission autosomique dominante: maladie de Thomsen.
Surtout les garçons qui sont plus sévèrement atteints. Début souvent précoce. Myotonie diffuse.
4.2. Canalopathies sodium.
Paramyotonie congénitale d’Eulenburg et adynamie périodique héréditaire de Gamstorp.
Accès paramyotoniques suivis ou non de fatigue généralisée débutant dans l’enfance.
Maladie de Gamstorp: accès paralytiques rapidement résolutifs de fréquence décroissant avec l’âge. Fréquence maximale entre 15 et 25 ans. Les accès sont déclenchés par un court repos après un exercice
important. Peuvent être déclenchés par le froid ou le jeûne glucidique. Entre les accès la myotonie est pratiquement constante.
5. AUTRES DYSTROPHIES MUSCULAIRES.
La classification s’est appuyée sur la connaissance génétique et biochimique des protéines contractiles du muscle strié.
5.1. Dystrophie musculaire autosomique récessive sévère de l’enfant (SCARMD).
Haute prévalence dans le Maghreb. S’exprime dans les deux sexes. Hétérogénéité génétique.
5.2. Dystrophies musculaires congénitales. (Howard, 1908) (CMD)
Hérédité autosomique récessive. Souvent remarquées par une diminution des mouvements foetaux intra-utérins.
Dystrophie musculaire congénitale occidentale (CMD type I): arthrorogrypose néonatale. Hypotonie néonatale sévère.
Dystrophie musculaire congénitale type Fukuyama (type II): atteinte associé du système nerveux central, convulsions, hydrocéphalie, retard mental.
Autres formes associées à d’autres malformations (squelette, cornée...)
5.3. Dystrophie Facio-Scapulo-Humérale.
Déjerine-Landouzy (1884). Autosomale dominante. Début à la face. Relativement fréquente mais souvent méconnue. Début dans l’adolescence, évolution très lente.
5.4. Myopathies “des ceintures”.
Myopathie des ceintures : autosomale récessive, cas sporadiques très nombreux. début deuxième ou troisième décennie.
Myopathie quadricipitale. Diagnostic difficile avec certaines myosites à inclusions (biopsie).
5.5. Myopathies oculo-pharyngées. (OPMD)
Début tardif après 50 ans. Autosomique dominant. Ptosis, dysphagie d’installation progressive.
5.6. Dystrophie musculaire d’Emery-Dreifuss (EDMD).
Récessive liée à l’X. Dystrophie huméro-péronière avec rétractions associée à des troubles de conduction cardiaque. Les rétractions tendineuses sont souvent précoces.
5.7 Dystrophies scapulo-péronières.
Age variable. maladie de “Stark Kaeser”.
5.8 Myopathies distales
Myopathie distale (Gowers - 1902).
Formes à début précoce ou tardif. Age de début très variable.
F. LES SOINS INFIRMIERS
En l'absence de thérapeutique étiologique, les soins infirmiers sont absolument capitaux bien que particulièrement ingrats, devant des malades allant s'aggravant inexorablement.
|
